

Protein space and the emergence of proteins on primordial Earth
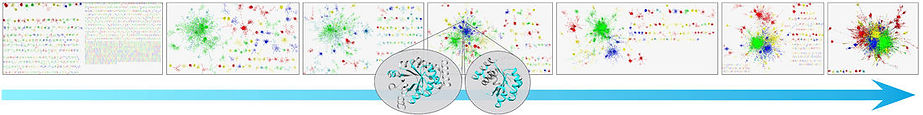
Proteins share similar amino acid segments with one another. Such 'reuse of parts' has clear evolutionary advantage over ‘design from scratch', which often generates segments that cannot even fold, let alone be active. The best characterized form of reuse involves ‘domains’, where the shared segments are of 100 amino acids on average. It is, however, conceivable, that proteins reuse even smaller segments. If so, such segments can be regarded as the basic evolutionary units of proteins. To study this eventuality, we searched among two large and representative sets of proteins (~15,000 domains and ~20,000 full chains) for elongated amino acid segments that are similar in sequence and structure. Using an in house methodology that was tailor made for this study, we presented the results as a similarity network, where the proteins/domains were shown as nodes, and the edges connected proteins/domains that are similar, i.e., evolutionarily related. Indeed, this network view showed that protein space features a large connected component of domains that share smaller segments with each other, as well as many isolated ‘islands’, each corresponding to specific architecture (see figure below). We observed reuse of amino acid segments of variable lengths, and called the reused parts ‘themes’ (as in ‘variations on a theme’). Very complex patterns emerged, where the same amino acid can appear both within a short reused theme, shared by many proteins, or within a longer theme, shared by few. This study suggests a recursive model of protein evolution, in which themes of various lengths, typically smaller than domains, ‘hop’ between contexts, and those that are fit remain, leaving traces for us to detect.
.jpg)
Networks of similarity among protein structural domains. Edges connect nodes that correspond to similar domains, such as the two at the center, sharing similarity in the cyan segment. Considering networks based on increasingly lax thresholds to identify similarity relationships is analogous to moving back in evolutionary time. Image courtesy of Varda Wexler of the multimedia unit of the faculty of Life Sciences.
In another project we aimed to shed light on a key mystery in protein function evolution: how did protein-ligand binding and recognition emerged, and how have they continued to evolve? To study these questions we focused on proteins that interact with adenine-containing enzyme cofactors, such as ATP, flavin adenine dinucleotide (FAD), S-adenosyl methionine (SAM), etc. We chose these cofactors because they have been present on Earth since the beginning of life. We designed a computational pipeline, ComBind, which superimposed the structures of ~1,000 proteins based on their adenine-containing cofactors, and then compared the patterns of hydrogen bonds between protein and adenine. This analysis showed that, in contrast to previous suggestions, evolution has maximized protein-adenine binding interactions by using all three faces of adenine. Furthermore, we found that these interactions are mediated by short linear sequence motifs on the protein side. While such motifs have been previously identified, we significantly expanded the list of these motifs. As anticipated, homologous proteins shared similar adenine-binding motifs, but occasionally, proteins that do not share evolutionary origin, nevertheless shared similar adenine-binding motifs. Most interestingly, we found a link between adenine binding motifs and protein themes: specific themes are often involved in adenine binding (see figure on the right). By searching the UniProtKB database for these themes, we identified over a million proteins of unknown structure that are predicted to bind adenine. This is the first step in our attempt to establish a functional alphabet for proteins based on themes. Overall, our analysis highlighted the opportunistic nature of evolution and suggested that adenine binding emerged more than once. It offered a possible scenario for the emergence of protein function, where primordial peptides with minimal biological activity (e.g., ligand binding) gradually evolved into current proteins.

Membrane proteins

Membrane proteins fulfil key biological functions on the cellular and physiological levels, they are involved in numerous diseases, and accordingly, they constitute prime targets to pharmaceutical drugs. Studying membrane proteins is particularly challenging, especially on the structural level, because of their complex environment. Our research of membrane proteins has been focusing both on specific proteins and on principle aspects of protein-membrane interactions. Currently, we are mainly studying Cation/Proton Antiporters (CPAs), which play a crucial role in maintaining cells pH levels and salt concentrations. With more than 100,000 entries in UniProt, CPAs comprise one of the largest protein superfamilies. We exploited evolutionary data to predict the functional traits of CPA. Because of the multitude of CPA sequence homologues, their evolutionary relationship is complex and challenging to reconstruct, but for the exact same reason it can be very informative. We conducted the most extensive evolutionary analysis of CPAs carried so far, encompassing ~6,500 protein homologs. The analysis identified an eight-amino acids sequence motif, which not only distinguishes the two main groups of CPAs, but also seems to determine both their electrogenicity (i.e., whether they pass one or two proton per each cation) and cation selectivity (Na+ vs. K+). Furthermore, the analysis provided a new way to classify CPAs, and showed that contrary to previous suggestions, the distinction between the two main CPA groups only partially correlates with their electrogenicity. This allowed us to identify specificity-determining residues in this family (see figure on the right), and based on this, to modify CPAs function and also recover activity in an inactive CPA mutant (both verified by the Padan laboratory, HUJI). We intend to further investigate these interesting transporters using simulations and other computational techniques, where our long term goal will be to provide a detailed atomic model of the transport mechanism.
ConSurf
Jointly with the Mayrose and Pupko labs, we have been developing ConSurf, an automated tool that accurately predicts the evolutionary conservation of amino acid positions in proteins, and also presents the conservation on the structures of these proteins as a color map (see figure on the right). This type of analysis is of great importance to biologists, as protein positions that are functionally important tend to change much more slowly during evolution compared to those that do not have specific function. Thus, ConSurf can help protein scientists to identify functional positions in new proteins. It can also assist protein engineers finding positions within proteins that can be safely mutated to increase their activity or to change their specificity towards their substrates. While there are other methods that calculate the evolutionary conservation of proteins, ConSurf does it very accurately because it uses statistically robust approaches (Bayesian or maximum likelihood).

Rational Drug Design
Traditional high throughput screening is a laborious and costly research, which typically is out of the scope of the academy. We use computational methods to screen large combinatorial libraries for novel drug candidates. The search algorithm finds compounds with good stereochemical fit to the structure of the protein binding site.
An example is our collaboration with the Azem Lab, where we target the Hsp60 chaperonin, which is implicated in multiple cancers. The Azem lab has determined the structure of Hsp60 in complex with its co-chaperonin Hsp10, obtaining a complex with a football-like shape. We screened a database of several million compounds, fitting each compound to the nucleotide binding site of Hsp60 and calculating the binding energies. The different compounds were ranked according to that energy. From the top ranking compounds we selected 10 compounds to be tested in empirical binding assays.
